Chemissian
Chemissian is a powerful tool for analyzing the electronic structure and spectra of molecules. With this tool, you can build and analyze molecular orbital energy-level diagrams using both Hartree-Fock and Kohn-Sham methods. Additionally, you can compare calculated experimental UV-VIS electronic spectra with experimental ones on the same plot, calculate and visualize spin natural orbitals, natural transition orbitals, electronic and spin densities, and prepare them for publication. The graphical interface of Chemissian is user-friendly, making it easy to examine and visualize data from output files generated by various quantum chemical program packages, including Gaussian, US-Gamess, Firefly/PC-Gamess, Q-Chem, Molpro, NWChem, ORCA, Turbomole and Spartan. Chemissian can be used to investigate the nature of transitions in UV-vis spectra, as well as the bonding nature of molecules.
For the news see news page
Chemissian Features:
Build Molecular Orbitals energy level diagrams
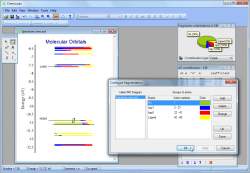
- With the integrated graphical editor, it's a breeze to add text labels to MO diagrams. You can also add connecting lines between MO energy levels and "occupy" the energy levels with electrons. This makes it much easier to visualize and understand the molecular orbitals:
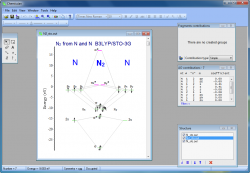
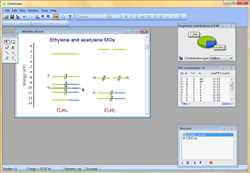
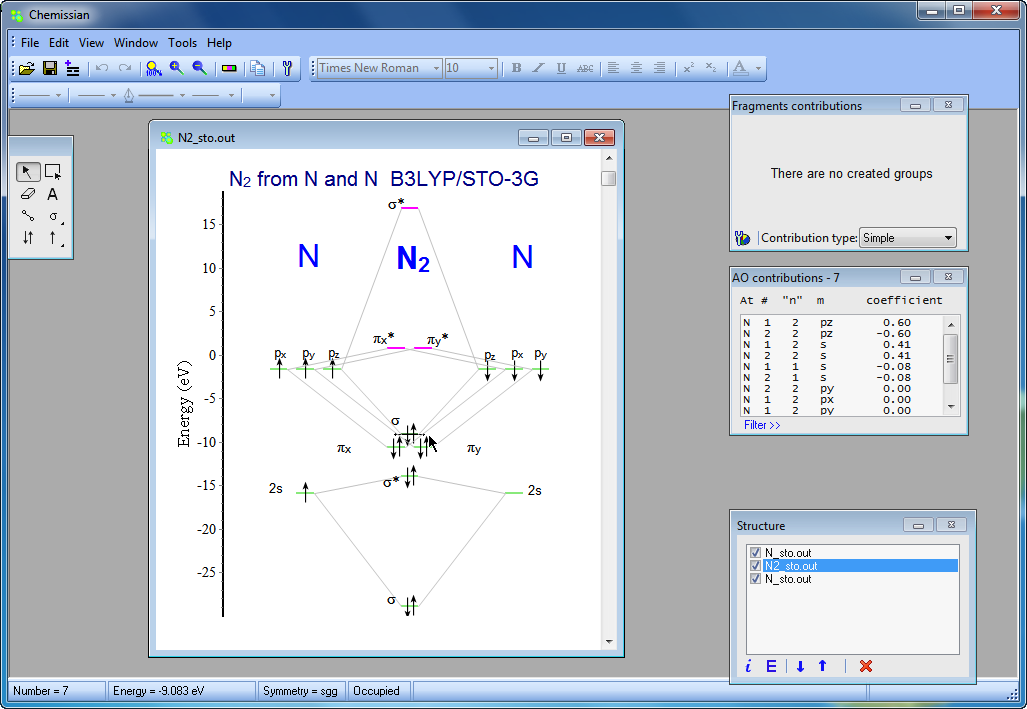

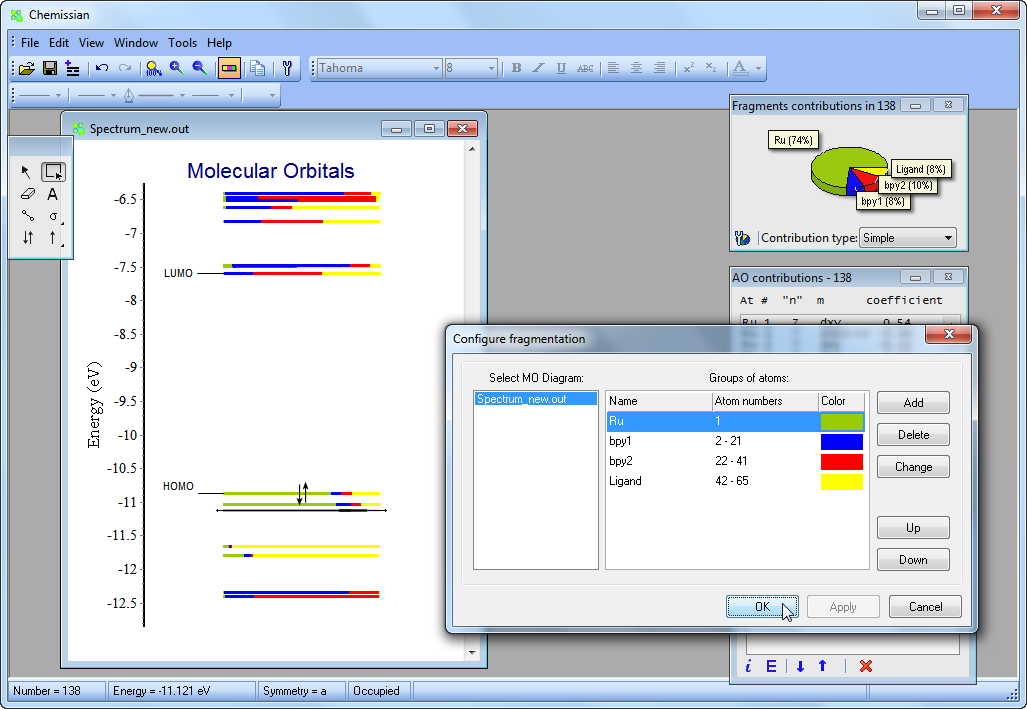
- Chemissian offers the ability to examine the electronic structure of molecules, including the option to navigate between energy levels using either a keyboard or mouse cursor. Additionally, Chemissian provides a clear visual representation of the contributions to the current molecular orbital from atomic or molecular fragments, presenting the information on either a contribution diagram or directly on the MOs diagram.:
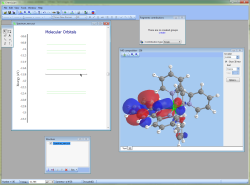
Build, visualize and interpret UV-Visible Spectra from Gamess, Firefly, Gaussian, Spartan, Q-Chem, Orca and NWChem outputs
Chemissian provides a comprehensive graphical analyzer that can analyze the composition of MOs and electronic spectra of molecules. With a range of tools available, you can build electronic UV/VIS spectra from output files generated by various quantum-chemical programs such as GAMESS, Firefly(PC-GAMESS), GAUSSIAN, Spartan, NWChem, Orca or Q-Chem:
- It is possible to easily visualize a spectrum by simply loading the output of Gamess/Firefly/Gaussian/Q-Chem/Spartan/NWChem/Orca calculation which contains the TDDFT/CIS data.
- By displaying both experimental and calculated spectra on a single diagram with the same wavelength scale, you can make a convinient comparison.
- It is possible to include multiple spectra in one diagram, which can be particularly helpful when examining the impact of the solvent on the spectrum.
- Similar to the MO diagram editor, one can navigate through the peaks in a spectrum and link the present peak with the transitions happening between MO levels.
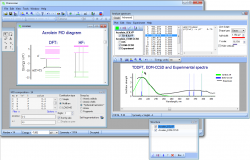
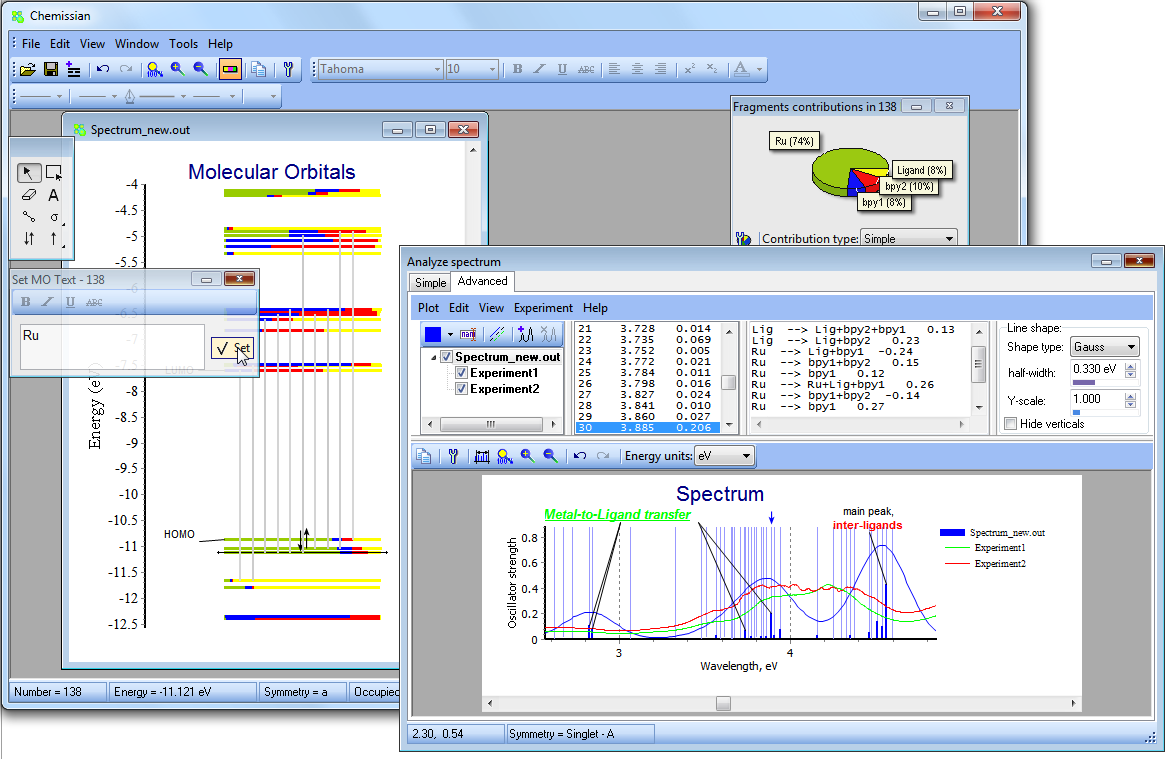

- In Chemissian, you can customize the spectrum diagram by including text labels and various graphical objects. You also have the option to use different energy units for your convenience.
- With the assistance of Chemissian tools, one can analyze and comprehend the nature of spectral transitions, such as metal-to-ligand charge transfer, ligand-to-ligand charge transfer, pi-pi* and more. This is achieved by utilizing the information of molecular orbitals composition extracted from the output files of various computational chemistry software, including Gamess, Firefly, Gaussian, Q-Chem, Spartan, Orca, and NWChem.
- Calculating and visualizing natural transition orbitals:
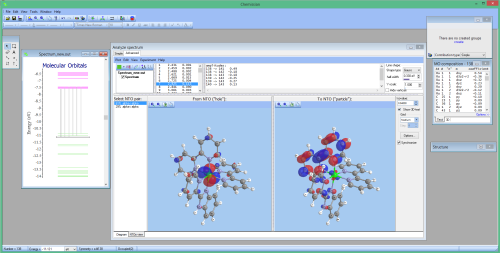

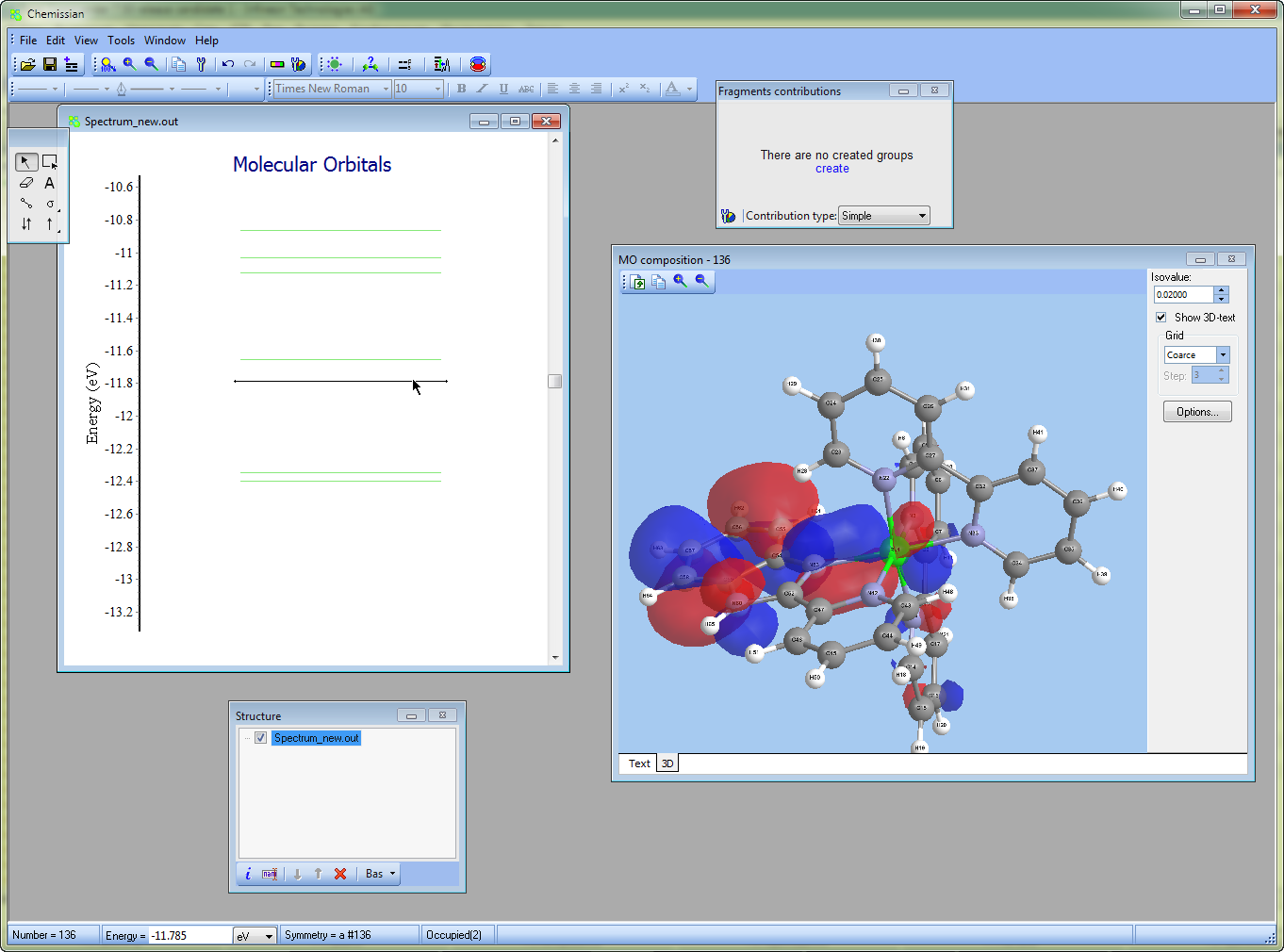
Analyze electronic density distribution
- With the help of Chemissian, one can examine the distribution of electronic and spin density, as well as the difference or "defomation" density. Additionally, individual molecular orbitals can be analyzed, along with any linear combination of these variables, which can be useful for plotting Fukui functions and other related tasks.
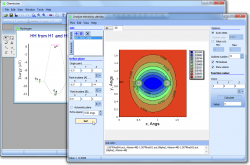

- Chemissian can build the distributions as three-dimensional surfaces,
- two-dimensional contour maps or
- build the distribution along a given line (one-dimensional).
- For this only the standard Gamess/Firefly/Gaussian/Q-Chem/Molpro(Molden)/Orca(Molden)/Spartan output file is used, e.g. no cube-files are required.
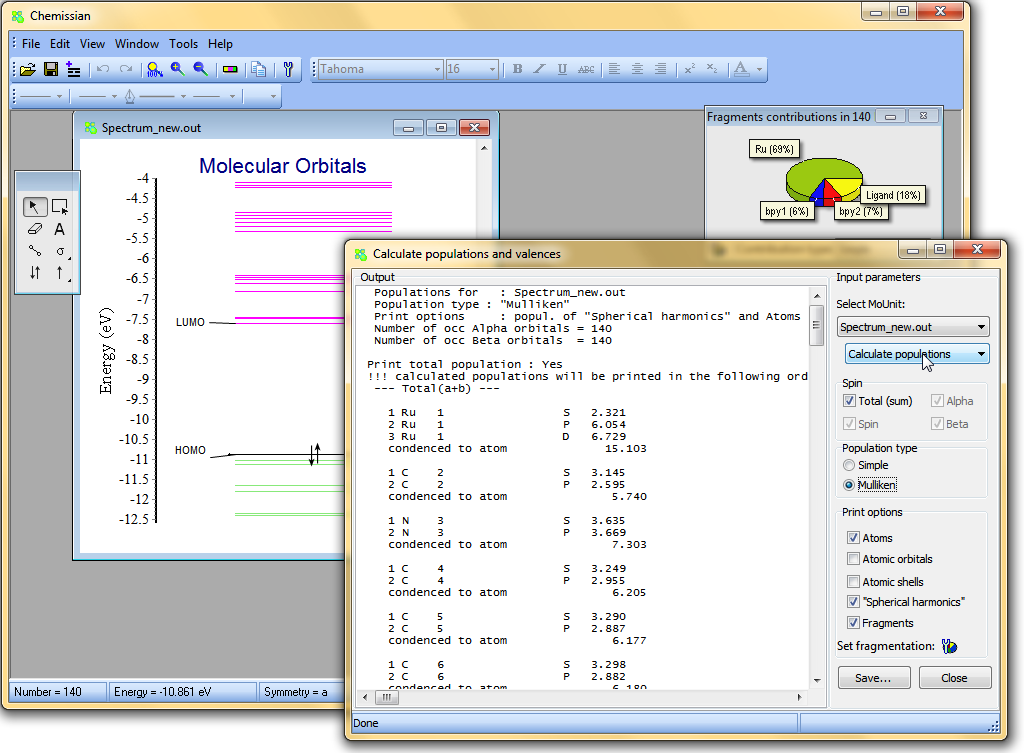
Calculate populations and valences
- Chemissian can calculate Mulliken and Simple populations of atomic orbitals, shells, "spherical Harmonics", atomic or molecular
fragments (any group of atoms):
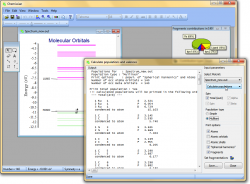
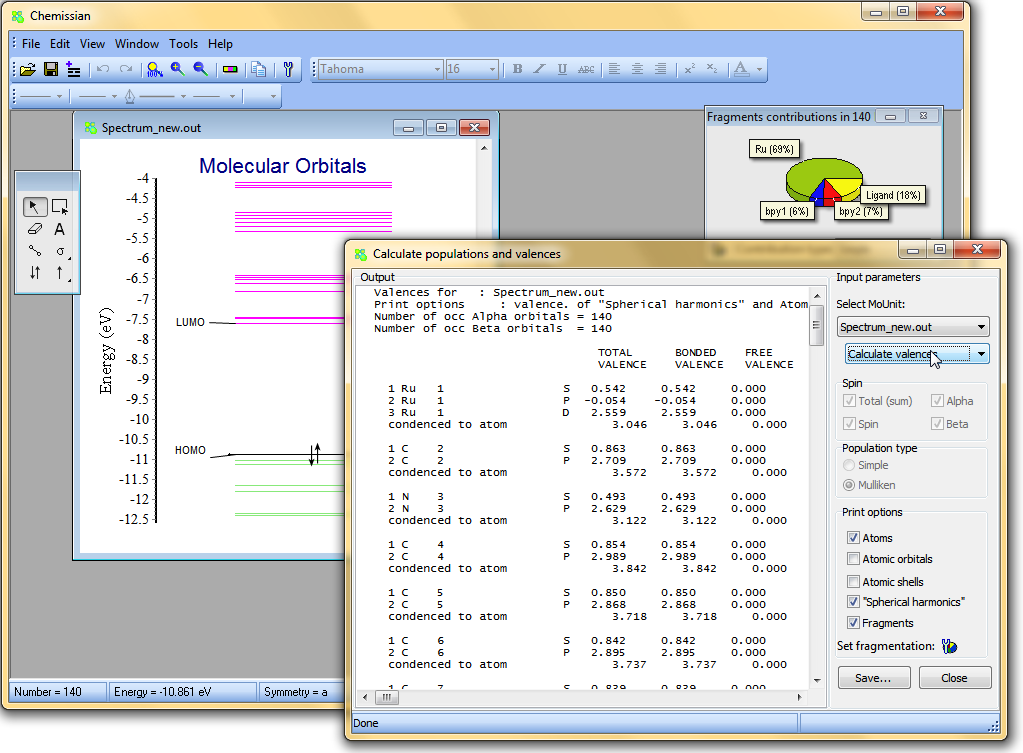
- It is possible to calculate valences of atomic orbitals, shells,"spherical harmonics", atoms and fragments:
- Analyze molecular orbital composition - calculate
contributions from atomic orbitals, atoms, molecular fragments, shells, etc. to MOs:

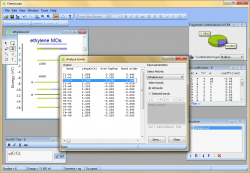
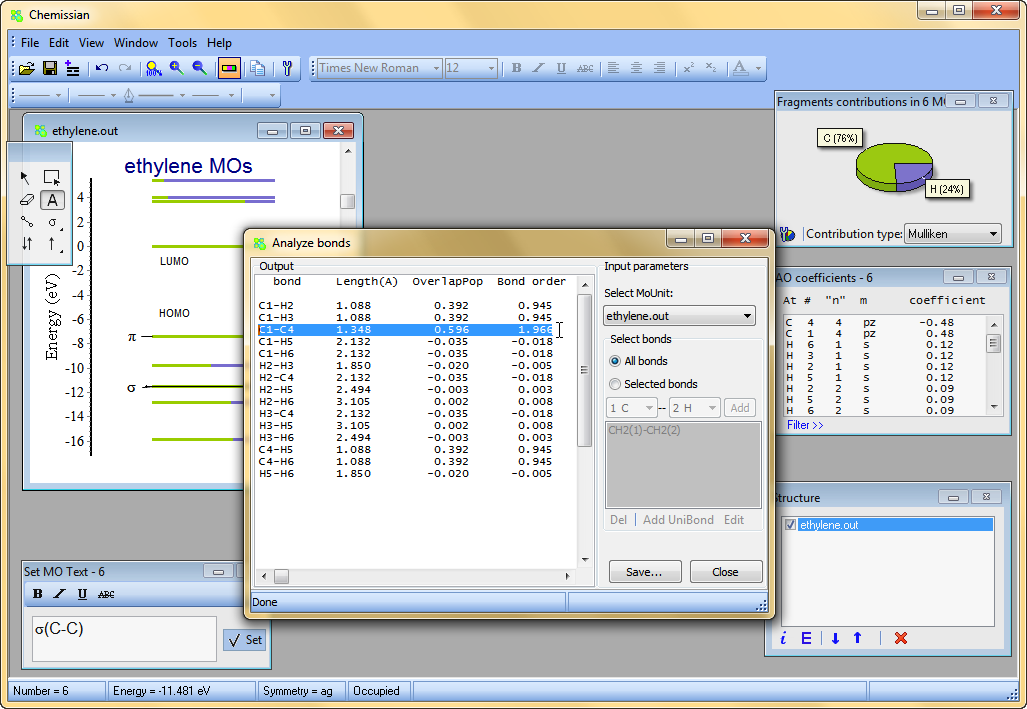
Calculate quantum-chemical bond order indexes and overlap populations
- Use Chemissian to investigate the bonding nature in molecules - calculate quantum-chemical bond order indexes and overlap populations for every bond in a molecule. It is also possible to analyze "generalized bond, e.g. "bond" between molecular fragments.
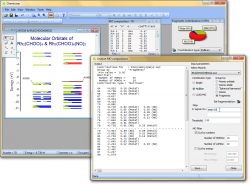
Work with several calculations at the same time
It is possible to gather and analyze the results of multiple calculations in one document using Chemissian. For instance, if you have the starting reagents and the final reaction product, you may want to examine the changes in the electronic structure. To do so, you can add multiple calculations onto a single diagram, and they will be displayed on a shared energy scale. This allows you to switch between different calculations and compare the electronic structures of all the participants simultaneously.
Save results in a single file
Save the obtained document in a Chemissian file format. It will allow one to keep all data in a single compressed file (uncompressed wave function takes up a lot of disk space); at any time it is possible to open and proceed to work with a saved document, analyze, edit the data, share with colleagues, etc.